Geometrical Analysis of Genome for COVID-19 vs. SARS-Like Viruses
Mads Bahrami
Wolfram Research, Inc.
Z-curve theory provides a very unique geometrical approach for analyzing a genetic sequence, while preserving all the genome information. In this computational essay, I have developed a very efficient and fast function (in the Wolfram Language) to generate the Z-curve of any genetic sequence, regardless of its length. Then I generate Z-curves of 39 COVID-19 viruses together with 40 SARS-like viruses, using their complete genomes. Numerical analysis of corresponding Z-curves and their clustering shows a very close phylogenetic relationship between the family of COVID-19 viruses and the bat coronavirus isolate RaTG13 (MN996532), therefore supporting the hypothesis of a bat origin for COVID-19.
Collecting Data
Here is the accession list of SARS-like viruses:
viruses="AY274119.3","AY278488.2",
SequenceIcon
,"NC_004718","NC_009019";
One can use the function ImportFASTA, from the Wolfram Function Repository, to import FASTA data from the NCBI using an NCBI Reference Sequence. To save time, I have done this and given it an iconized form.
In[]:=
virusNameSeq=
VirusNameSequence
(*Map[ResourceFunction["ImportFASTA"],viruses]*);
Take the first element from all sublists of virusNameSeq, flatten the result and return the substrings from the first space to ",":
From the previous imported data, take only the Accession and Sequence columns, make them an association with accessions as keys and sequences as values, then replace the sequences using srules:
Now let us find the three nearest Z-curves of SARS-like viruses to the Z-curve of each COVID-19 virus. Without loss of fidelity and to compute faster, we will first project the Z-curves into 30-D.
Reduce the dimension to 30-D:
Find the three nearest Z-curves of SARS-like viruses to the Z-curve of each COVID-19 virus:
Flatten out the closest and obtain tallies:
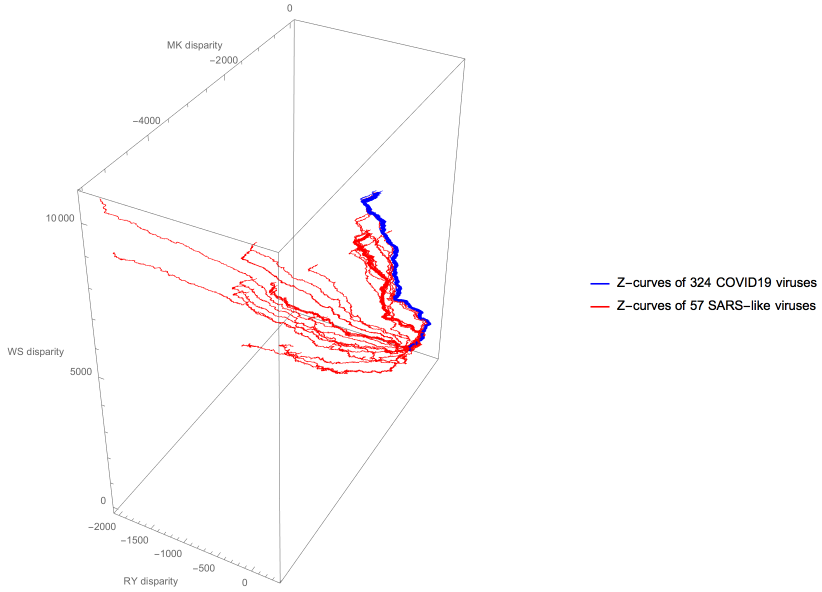
As suggested by the Z-curve visualization, the three nearest SARS-like viruses are the same for all COVID-19 viruses.
Delete duplicates from the previous result and return their GenBank titles:
Our analysis shows that among the 57 SARS-like viruses that we study here, bat coronavirus isolate RaTG13, bat SARS-like coronavirus isolate bat-SL-CoVZXC21 and bat SARS-like coronavirus isolate bat-SL-CoVZC45 have the closest genome similarity to the COVID-19 viruses, providing evidence for a bat origin of COVID-19.
Let us do some hierarchical clustering.
Cluster the viruses based on their Z-curves using a single-linkage clustering algorithm:
Assign a different color to each cluster, and represent 3D images of corresponding Z-curves:
Project clusters into 3D using the t-distributed stochastic neighbor embedding algorithm:
Construct a dendrogram from the hierarchical clustering of the Z-curves (note the coloring is the same as the one for clusters; those that are bold and italic are SARS-like viruses):
Concluding Remarks
I generated the Z-curves of 39 available genetic sequences of COVID-19 viruses together with 40 SARS-like viruses. An eyeballing of the 3D visualization of the Z-curves suggests that there are three SARS-like viruses very close to the virus family of COVID-19. By qualifying the similarity/dissimilarity between Z-curves, I identify those three viruses as bat coronavirus isolate RaTG13 (MN996532), bat SARS-like coronavirus isolate bat-SL-CoVZXC21 (MG772934 .1) and bat SARS-like coronavirus isolate bat-SL-CoVZC45 (MG772933 .1). The hierarchical clustering of viruses based on the Z-curves also confirms the visualization, and identifies bat coronavirus isolate RaTG13 as the most likely culprit of COVID-19. My results strongly support the hypothesis of a bat origin for COVID-19.
Acknowledgments
I would like to thank Dr. Robert Nachbar and Dr. Daniel Lichtblau for their valuable feedback.
References
Zhang, Ren and Chun-Ting Zhang. 2014. “A Brief Review: The Z-Curve Theory and Its Application in Genome Analysis.” Current Genomics 15(2) (April): 78–94. https://doi.org/10.2174/1389202915999140328162433.
Zheng, W. X., L. L. Chen, H. Y. Ou, F. Gau and C. T. Zhang. 2005. “Coronavirus Phylogeny Based on a Geometric Approach.” Molecular Phylogenetics and Evolution 36(2) (August): 224–32. https://doi.org/10.1016/j.ympev.2005.03.030.